
BIOVIA COSMO-RS
A Comprehensive Toolbox for the Modelling and Prediction of Fluid Phase Properties
Predict Solutions Faster and More Accurately
COSMO-RS simulations enable chemical engineers, chemists, formulation engineers and materials scientists to research and develop new solutions faster and more efficiently than with test and experimentation alone, thus accelerating innovation and reducing costs.
COSMO-RS simulations are based on a sound scientific theory, which ensures robust and reliable predictions over the whole range of chemistry in the liquid state. The first principle approach allows for predictions of new, not yet synthesized compounds, reaching beyond the known chemical space. BIOVIA’s COSMO team consist of the original inventors of COSMO-RS, assuring timely support and prime expertise to help solve even the most challenging problems in solution thermodynamics.
Key Benefits
- A robust scientific foundation combining quantum chemistry and thermodynamics ensures accuracy and reliability
- Reliable predictive capabilities help companies explore new areas with confidence
- A broad range of applications, properties and solutions enable adaptation to diverse needs, from optimizing formulations to discovering new materials
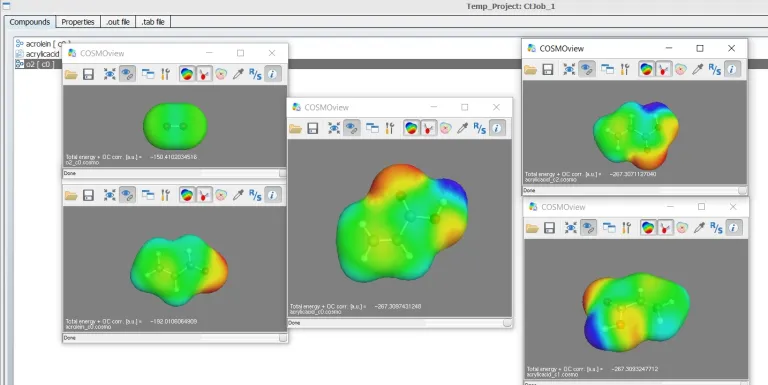
COSMO-RS Products

Efficient Treatment of Self-organizing Systems and Biomembranes

The Shortcut to COSMO-RS

Advanced COSMO-RS Implementation with Quantum Chemically Generated Charge Density Surfaces
Start Your Journey
The world of product development is changing. Discover how to stay a step ahead with BIOVIA.
FAQ About COSMO-RS Solutions
By providing fast and reliable predictions based on a sound scientific theory, COSMO-RS enables chemical engineers and scientists in the chemicals industry to develop new compounds more efficiently. It reduces time and costs associated with traditional testing methods, enhancing data collection and supporting digital transformation in chemical research.
Industries involved in chemical engineering, materials science, formulation development, and drug design benefit significantly from COSMO-RS simulations. For chemical manufacturers, the tool provides predictive capabilities that go beyond traditional experimental methods, improving data collection and accelerating digital transformation efforts.
COSMO-RS uses a first-principle approach, allowing it to predict properties of new, unsynthesized compounds accurately, extending the scope of research beyond known chemical spaces.
While COSMO-RS simulations significantly reduce the need for physical experimentation by providing accurate predictive data, they are often used in combination with experimental data collection. This approach refines and optimizes results, helping chemical manufacturers implement digital transformation in their research processes.
Also Discover

Solutions for Research, Development, Quality, and Manufacturing

Foster Innovation with In Silico Design

Fast and Robust Quantum Chemistry
An Integrated, Multi-scale Modeling Environment
Learn What BIOVIA Can Do for You
Speak with a BIOVIA expert to learn how our solutions enable seamless collaboration and sustainable innovation at organizations of every size.
Get Started
Courses and classes are available for students, academia, professionals and companies. Find the right BIOVIA training for you.
Get Help
Find information on software & hardware certification, software downloads, user documentation, support contact and services offering